
Could Genomics Influence Prostate Cancer Treatment?
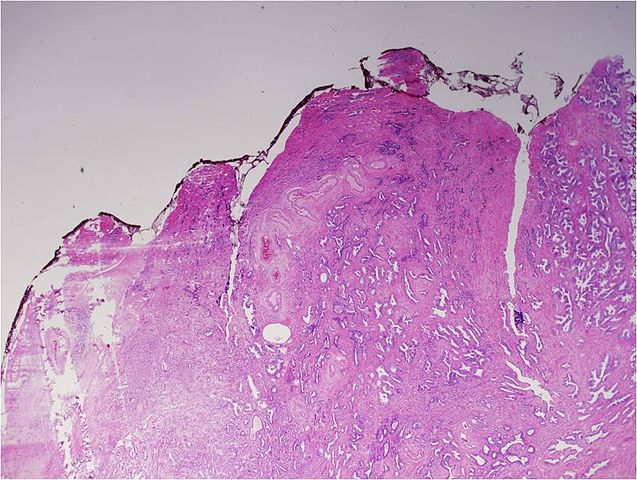
Gleason pattern 3. Prostate cancers composed entirely of Gleason pattern 3 are given a Gleason score of 6. Image by Difu Wu.
Prostate cancer is one of the most common cancers diagnosed in the United States. It is estimated that it will account for 9.6 percent of the 1,688,780 new cancer cases diagnosed in the country this year, which translates into 161,360 new cases of the disease.
About one-third of men diagnosed with prostate cancer have a tumor that grows very slowly, making it very unlikely that the disease will progress to become a clinical problem. As Johann S. de Bono, MD, PhD, highlighted in a recent post on this blog, characterizing new ways to identify these cancers is an area of active research investigation because “… it would be ideal if we could decrease the overtreatment of benign disease and focus on improving the treatment of aggressive prostate cancers.”
New insight into this issue was provided recently in a paper published in Cancer Research, a journal of the American Association for Cancer Research, in which researchers reported a potential new genetic biomarker to identify patients with prostate cancer with a low risk of spreading.
Assessing risk for prostate cancer progression
The National Comprehensive Cancer Network (NCCN) is an alliance of 27 U.S. cancer centers that work together to develop treatment guidelines for most cancers. For prostate cancer, current NCCN guidelines recommend different treatment approaches depending on a patient’s risk for disease progression and death from prostate cancer. Among the factors affecting a patient’s risk for disease progression and death from prostate cancer is the Gleason score assigned to their tumor.
A Gleason score is calculated after analysis of the patient’s prostate needle biopsy specimen under a microscope. Depending on how normal or abnormal the tumor looks, it is assigned a grade from 1 to 5, with 5 being the most abnormal and most aggressive. Different areas of a tumor may have different patterns, and the Gleason score is calculated by adding together the predominant grade and the highest nonpredominant grade.
Prostate cancers with a Gleason score of 6 are considered to have a very low risk or low risk of disease progression, depending on other factors such as the number of biopsy cores in which cancer was detected. NCCN guidelines recommend active surveillance as an option for men with very-low risk or low-risk prostate cancer. Active surveillance entails regular testing followed by curative treatment if the cancer progresses.
Up to 50 percent of men with prostate cancer who choose active surveillance ultimately have disease progression and require curative treatment.

John C. Cheville, MD
“Identifying a biomarker that, in addition to Gleason score, distinguishes men at increased risk for disease progression from those whose prostate cancer never becomes a clinically significant problem could help improve patient care,” said the lead author of the recent Cancer Research paper John C. Cheville, MD, professor of pathology at the Mayo Clinic in Rochester, Minnesota.
Can genomics help define risk for prostate cancer progression?
To look for genetic biomarkers of clinically significant or insignificant disease, Cheville and colleagues used whole-genome mate pair sequencing to study gene fusions in prostate cancer tissue samples obtained from 133 patients who underwent a radical prostatectomy at the Mayo Clinic. The researchers focused on fusions between the TMPRSS2 gene and the ETS family of transcription factors because they occur in about 50 percent of prostate cancers, with TMPRSS2-ERG gene fusions being the most common.
The prostate cancer samples in the study were divided into four groups: 53 low volume Gleason 6 tumors were classed as very low risk for progression; 26 high volume Gleason 6 tumors were classed as low risk for progression; 29 Gleason 7 tumors were classed as intermediate risk for progression; and 25 Gleason 8 or higher tumors were classed as high risk for progression.
TMPRSS2-ERG fusions were detected in 45 percent of all the prostate cancers analyzed. However, the frequency of the fusions in the different groups varied: Fusions were detected in 43 percent, 49 percent, 52 percent, and 24 percent in the very-low risk, low-risk, intermediate-risk, and high-risk groups, respectively.
When the researchers examined how the fusion formed—deletion of the interstitial genes or chromosomal translocation with interstitial gene retention—they found that among the 60 prostate cancers with TMPRSS2-ERG fusions, 39 had deleted the interstitial genes between TMPRSS2 and ERG during the fusion event and 21 had retained these genes. Eighteen of the 21 prostate cancers that had retained the interstitial genes during TMPRSS2-ERG gene fusion were in the very-low risk and low-risk groups.
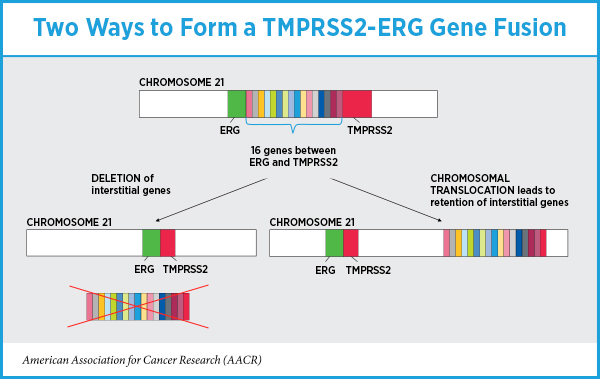 “Our data support results from other studies in that the presence or absence of a TMPRSS2-ERG gene fusion was not predictive of outcome,” Cheville said. “But how the gene fusion formed was important; the retention of interstitial genes during the fusion event was more common in very- low risk and low-risk cancers, and there may be genes in this region that suppress or limit tumor growth.”
“Our data support results from other studies in that the presence or absence of a TMPRSS2-ERG gene fusion was not predictive of outcome,” Cheville said. “But how the gene fusion formed was important; the retention of interstitial genes during the fusion event was more common in very- low risk and low-risk cancers, and there may be genes in this region that suppress or limit tumor growth.”
Information on whether a patient went on to have a biochemical recurrence was available for 34 patients who had prostate cancer with a TMPRSS2-ERG gene fusion with interstitial gene deletion and 22 patients who had prostate cancer with a TMPRSS2-ERG gene fusion with interstitial gene retention. In univariate, but not multivariate, analysis, biochemical recurrence was significantly lower if the prostate cancer had a TMPRSS2-ERG gene fusion with interstitial gene retention compared with those that had interstitial gene deletion.
“There is potential utility for determining the status of interstitial genes in stratifying men with prostate cancer into more well-defined risk groups, but this will require further study before it can be incorporated into clinical practice,” concluded Cheville.
How else might genomics influence prostate cancer treatment?
A recent paper in another AACR journal, Cancer Discovery, suggests that genomic characterization of prostate cancer samples from African-American men has the potential to lead to the discovery of new investigational therapeutic targets because most large-scale genomic studies of prostate cancer have predominantly included samples from non-Hispanic white men.
In the study, exome sequencing of 102 samples from African-American men with localized primary hormone-naïve prostate cancer led to the identification of several gene mutations not previously observed in prostate cancer.
As highlighted by the authors, “Our results suggest that inclusion of sufficient numbers of patients of African ancestry in cancer genomic studies may enable the discovery of new cancer genes and inform the inclusion of diverse populations toward precision cancer medicine.”



Fine description