Molecular Targets 2021: Examining Synthetic Lethality Beyond PARP Inhibitors
Cancer is a very complex disease. Cancer cells often have the ability to rewire some of their functions to survive in harsh environmental conditions and bypass the toxic effect of therapies. But, paradoxically, some of the alterations that make them resilient can also leave them vulnerable.
In the past decade, researchers have advanced a novel anticancer therapeutic approach that targets specific proteins or pathways upon which cancer cells become dependent for survival as a result of genetic alterations.
This concept, called synthetic lethality, exists in nature, and was first discovered in fruit flies when researchers observed mutations that were compatible with life if they occurred individually but led to cell death when they existed at the same time.
The first approved synthetic lethal therapy, specifically poly (ADP-ribose) polymerase (PARP) inhibitors, targets DNA damage repair.
Building on the success of PARP inhibitors, researchers are now exploring other opportunities of synthetic lethality to develop a new generation of anticancer therapeutics, some of which are in clinical development. These were the focus of a session at the AACR-NCI-EORTC Virtual International Conference on Molecular Targets and Cancer Therapeutics, held earlier this fall.
Why target the DNA damage response?
Cells are equipped with a number of mechanisms to detect and repair DNA damage, which are finely orchestrated to preserve genomic integrity. The different DNA damage response (DDR) pathways and the major players implicated are extensively reviewed in this article from Cancer Discovery.
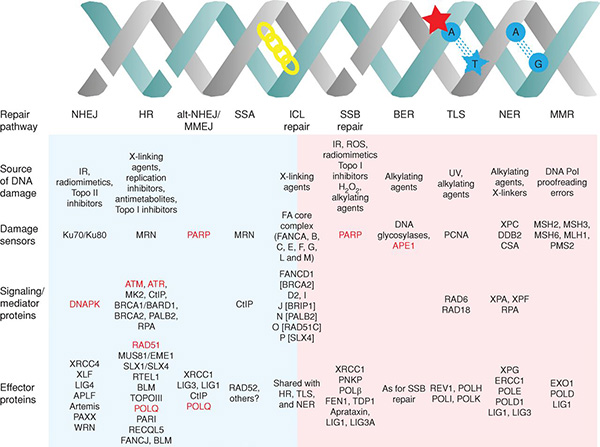
The interplay that exists between major DDR pathways creates dependencies whereby the functional loss of DDR components in tumor cells leads to reliance on other DDR mechanisms for survival following DNA damage.
Presenter Ruth Plummer, PhD, professor at Newcastle University, United Kingdom, noted how the discovery of the potential of synthetic lethality for cancer therapy was somewhat serendipitous.
Initially, DNA damage repair inhibitors were developed in an attempt to potentiate the effect of DNA-damaging treatments such as chemotherapy and radiation therapy. Disrupting the cells’ ability to repair DNA was expected to enhance the therapeutic effect.
The first-in-class PARP inhibitor rucaparib went into the clinic in 2003 in combination with chemotherapy.
In 2005, however, two seminal studies showed that PARP inhibition selectively killed cells with mutations in the BRCA1/2 genes, suggesting the possible use of PARP inhibitors as single agents in selected patients.
Homologous recombination (HR) is one of the main DDR pathways, specifically required to fix DNA double-strand breaks. Since tumors with mutant BRCA1/2 have defective HR, blocking an additional DDR pathway operated by PARP called base excision repair (BER) causes the accumulation of a critical amount of DNA damage that kills the cells.
“This is when the world changed,” Plummer said.
After successful clinical trials, the first PARP inhibitor olaparib (Lynparza) was approved in 2014 as a treatment for advanced ovarian cancer with BRCA mutations. Three more have since been approved (rucaparib, talazoparib, and niraparib) for certain ovarian, breast, and pancreatic cancers.
Beyond PARP inhibitors
“Now that PARP inhibitors are approved for use in the clinic, there are still multiple opportunities to build on their success,” said presenter Timothy Yap, PhD, associate professor at The University of Texas MD Anderson Cancer Center. “Not all patients with mutated BRCA1/2 respond to this therapy, and even if they do, drug resistance is nearly inevitable. There is still potential to deepen the responses, increase their durability, and expand the application of DDR inhibitors beyond currently approved indications.”
The DDR inhibitor landscape is rapidly expanding beyond PARP, accelerated by the discovery of new synthetic lethal interactions and precision targets enabled by cancer genome sequencing and the CRISPR technology.

POLθ inhibition
Presenter Geoffrey Shapiro, MD, PhD, professor at Harvard Medical School and institute physician at Dana-Farber Cancer Institute, discussed targeting the microhomology-mediated end joining (MMEJ) pathway, an error-prone repair mechanism for double-strand breaks that has been more recently identified.
BRCA-deficient ovarian, breast, and uterine cells with HR deficiency become dependent on MMEJ by upregulating the key enzyme of the pathway called polymerase theta (POLθ). Furthermore, since PARP is required to facilitate recruitment of POLθ at the sites of DNA damage, this represents an additional mechanism underlying the synthetic lethality of PARP inhibition in HR-deficient cells.
Shapiro and collaborators have identified the antibiotic novobiocin as a first-in-class POLθ inhibitor that demonstrated activity in BRCA-deficient preclinical models. Novobiocin has been included in the National Cancer Institute’s Experimental Therapeutics Program (NExT) for the development of clinical trials, to be tested as a monotherapy and in combination with PARP inhibitors.
Importantly, in cells that acquire PARP inhibitor resistance and partially restore HR despite BRCA deficiency, POLθ expression levels remain high and the protein is needed for DNA repair, suggesting that POLθ inhibition may reverse PARP inhibitor resistance and, therefore, be useful both in PARP inhibitor-sensitive and resistant settings.
The levels of POLθ mRNA and protein expression may be used as predictive biomarkers of response to novobiocin in PARP inhibitor-resistant cells, Shapiro said.
Shapiro also discussed the interaction of novel DDR inhibitors with the tumor immune microenvironment.
ATR inhibition
ATR, ATM, and DNA-PK are signaling kinases that sense DNA damage and are the master activators of the DDR. ATR, in particular, is important in the response to DNA replication stress, or replicative stress, which is caused by different insults including oncogene activation, chemotherapy, and radiation therapy.
Several ATR inhibitors are currently in phase I and II clinical testing, the most advanced being berzosertib and ceralasertib. These are being assessed as single agents or in combination with chemotherapy or other DDR agents, Yap noted.
Cancer cells that have lost ATM function become dependent on the ATR pathway and can be killed with ATR inhibition, suggesting the existence of a synthetic lethal relationship between ATR and ATM; however, ATR inhibition may be compensated by DNA-PK function.
Shapiro and colleagues have suggested that combining ATR and DNA-PK inhibitors may represent a more effective therapeutic strategy for patients harboring high levels of replicative stress or ATM inactivating mutations.
Yap discussed ATR inhibition in combination with PARP inhibition. ATR is required for the repair of stalled replication forks resulting from PARP inhibition, therefore, combined ATR and PARP blockage can lead to increased DNA damage and cell death. Multiple studies are currently ongoing to assess the effectiveness of this strategy.
In addition, ATR inhibition might be useful to overcome PARP inhibitor resistance, Yap added.
WEE1 inhibition
WEE1 is a kinase that regulates the cell cycle and participates in the ATR pathway. Yap reviewed different phase II clinical trials of adavosertib, a WEE1 inhibitor that is being tested in various tumor types with high levels of replicative stress and genetic instability—characteristics that are predicted to sensitize the tumors to WEE1 inhibition. These trials include uterine serous carcinoma, TP53 and KRAS-mutated metastatic colorectal cancer, and multiple tumor types with Cyclin E (CCNE1) amplification.
Although promising, adavosertib has adverse effects that can lead to dose interruption and, according to Yap, the newer, more selective WEE1 inhibitors might widen the therapeutic window.
The new kids on the block
Yap also discussed newer targets that have recently entered the clinic.
PKMYT1
PKMYT1 is a kinase that negatively regulates the G2/M transition of the cell cycle and was demonstrated to be synthetic lethal with CCNE1 amplification in a genome-wide CRISPR screening conducted by Repare Therapeutics. The highly selective PKMYT1 inhibitor RP-6306 is currently in phase I testing.
USP1
USP1 is a de-ubiquitinating enzyme that regulates the DDR in the Fanconi anemia pathway. A CRISPR screening platform by KSQ Therapeutics identified USP1 as a novel target on which cancer cells with DDR defects are dependent. Importantly, USP1 inhibition was more selective against cancer cells than other DDR inhibitors that were broadly cytotoxic. The first-in-class phase I trial of the KSQ-4279 USP1 inhibitor has just began and will test the drug as monotherapy and in combination with platinum-based chemotherapy and PARP inhibitors.
The importance of patient selection
“Patient identification is key to maximize benefits and minimize toxicities of DDR inhibitors,” Plummer said. “We have to find the right patient, the right tumor, and the right treatment.”
Next-generation CRISPR-enabled platforms for discovery of new synthetic lethal gene pairs allow for biomarker development at the preclinical stage and rational identification of the candidates before the start of clinical testing.
Tumor biology matters too, Plummer noted, as not all BRCA-related cancers behave in the same way. Studies have revealed different response rates to PARP inhibitors olaparib and rucaparib across different tumor types.
In addition, somatic mutations arising within the tumors contribute to response to DDR therapy. For example, in the ARIEL2 trial of rucaparib, similar efficacy was observed in patients with germline and somatic BRCA mutations, indicating that patient identification should not rely only on known germline genetics, but also on somatic mutations. Plummer added that we need ways to capture tumor mutations to make sure we identify most of the patients who would benefit from a treatment.
Somatic mutations are rare, however, so finding them might compare to the proverbial needle in a haystack, commented Plummer.
She suggested that circulating tumor DNA and real-time testing using blood-borne biomarkers might be key to overcoming these challenges and avoiding the difficulties and risks to the patients associated with tumor biopsies.