
Accelerated Approval of Cancer Drugs: How Many Verify Clinical Benefit?
Back in 1992, the U.S. Food and Drug Administration (FDA) established its Accelerated Approval Program so drugs that treat serious conditions and fill an unmet need could get to patients faster. This expedited approval is based on surrogate endpoints—such as a laboratory measurement, radiographic image, or physical sign—that the FDA says “is not itself a direct measurement of clinical benefit” but “is reasonably likely to predict clinical benefit.” That’s why these drugs must still be tested in a confirmatory trial that “shows that the drug actually provides a clinical benefit,” before it is granted regular approval.
But how many drugs granted accelerated approval subsequently confirm their clinical benefit? Are the ones that don’t subsequently pulled from the market? What happens if these trials show the drugs are beneficial, but for a different reason than what the accelerated approval was based on?
That’s what Edward Cliff, MBBS, MPH, Ian T. T. Liu, MD, JD, MPH, MS, and Aaron S. Kesselheim, MPH, MD, JD, at the Program On Regulation, Therapeutics And Law (PORTAL) within the Division of Pharmacoepidemiology and Pharmacoeconomics at Brigham and Women’s Hospital and Harvard Medical School, wanted to find out.
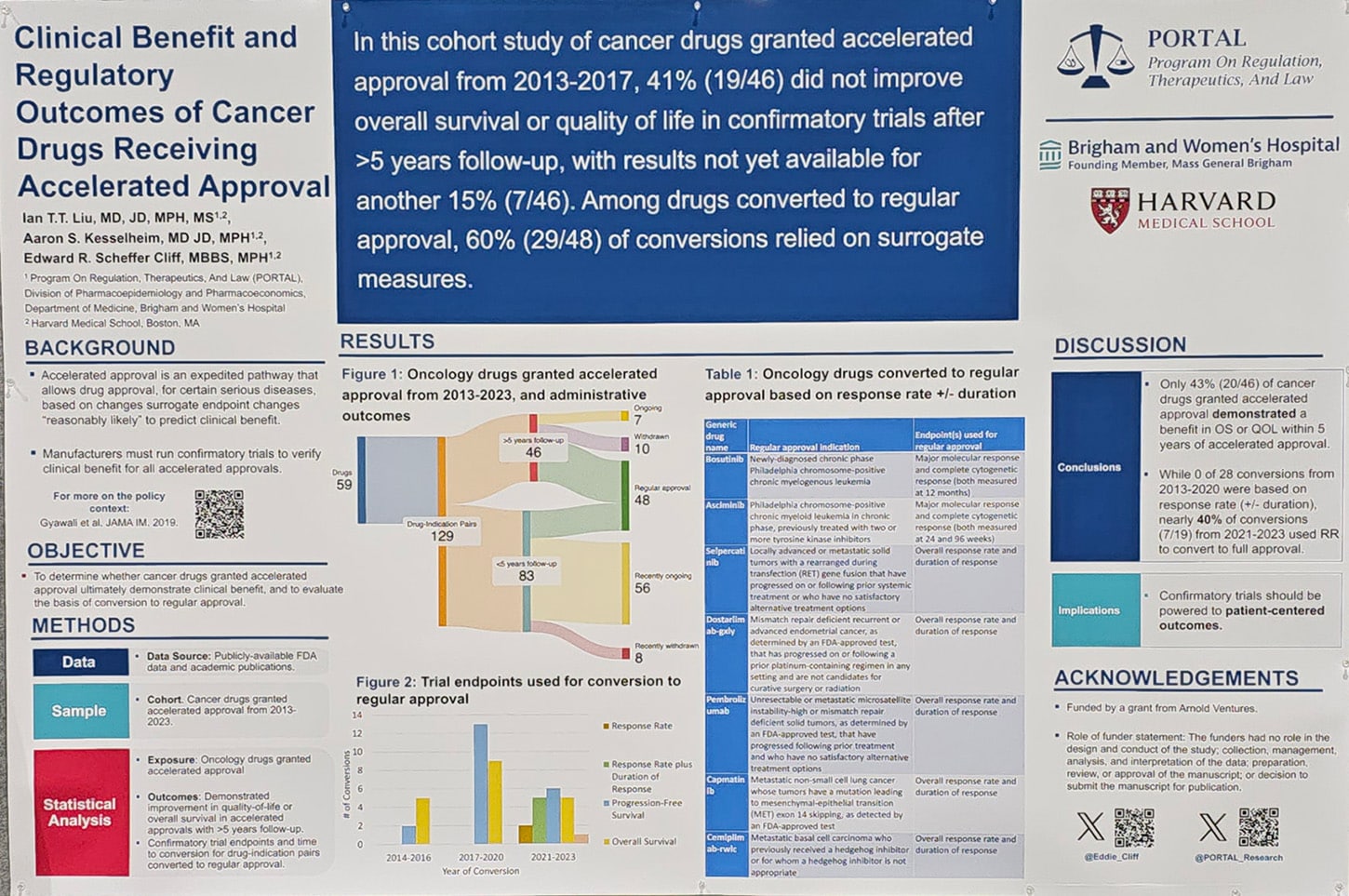
In a poster presented at the American Association for Cancer Research (AACR) Annual Meeting 2024, Cliff found that 59 cancer drugs were granted approval under the accelerated pathway between 2013-2023, which represented a total of 129 indications since some drugs were approved for multiple indications. Forty-six of those indications were approved between 2013-2017, giving them five years to conduct confirmatory trials. Of these, 43% demonstrated a clinical benefit and yet 63% were converted to regular approval.
“The FDA relinquishes significant leverage once it converts accelerated approvals to regular approval—it is harder to ensure timely completion of further trials, and harder to withdraw drugs—so the evidence used to justify these decisions is important,” Cliff said in a press release. “We found seven drugs that were converted to regular approval based on response rate, i.e., tumor shrinkage, but this leaves a great deal of uncertainty regarding whether the drugs ultimately benefit patients.”
What Endpoints Lead to Conversion to Regular Approval?
Out of the 129 cancer drug-indication pairs identified in the study that received accelerated approval, 48 were converted to regular approval—that includes some drugs that had less than five years to complete a confirmatory trial. By using publicly available data from the FDA, industry press releases, ClinicalTrials.gov, and peer-reviewed journal articles, the researchers found that the endpoints used to support conversion included progression-free survival (44%), overall survival (40%), response rate plus duration of response (10%), and response rate (4%). There was also a case in which a drug received regular approval even though the confirmatory trial showed negative results.
“We were surprised at the persistence of the finding of lack of evidence on clinical endpoints—these are drugs that are on the market and covered by insurance, so there’s no reason that they can’t be tested more commonly on meaningful clinical endpoints,” Liu said.
Cliff talked about the importance of using clinical endpoints that are appropriate for each tumor type. In highly lethal types of cancer—such as acute myeloid leukemia or glioblastoma—there should be emphasis on overall survival. In less aggressive tumor types where subsequent therapies can affect overall survival, he recommends a focus on collecting robust quality of life data to better inform whatever other endpoints are being measured. “You want to see multiple endpoints pointed in the right direction,” Cliff said.
How Many Accelerated Approvals Were Withdrawn?
Among the 46 drug-indication pairs that had at least five years for their confirmatory trials, 22% were withdrawn from the market while 15% still have ongoing trials after a median of 6.3 years. The researchers also found that during the period between 2013-2017, withdrawal time for a drug with accelerated approval that lacked clinical benefit decreased from 9.9 to 3.6 years. Further, conversion time to regular approval increased from 1.6 to 3.6 years.
“Faster, appropriate withdrawal decisions are a good thing for patients, as they ensure that ineffective drugs are on the market for a shorter period of time,” Liu said in the release. “While our study showed an increase in the time between accelerated approval and conversion to regular approval, we believe that conversion decisions should be both timely but—more importantly—supported by high-quality clinical outcomes, and that this is critical to the proper functioning of the accelerated approval pathway.”
How Many Accelerated Approvals Changed Indications?
When the researchers looked at whether the regular approval matched the same indication the accelerated approval was for, they found it changed in 62% of cases. Cliff said that in these instances, the regular approval was for an earlier line of therapy in the same cancer type in (38%), a broader indication without moving to an earlier line of therapy (17%), a narrowed indication (6%), and one changed in an alternate way.
“Whilst it is often beneficial to make promising new drugs available to more patients, running confirmatory trials in distinct populations from the initial pivotal trials risks leaving important clinical questions unanswered, which is particularly consequential if the confirmatory trial has a negative result,” Cliff said. Cliff and colleagues discuss the downsides of such approaches with examples in this article.
What Does This Mean for Physicians and Patients?
“We hope that these findings will encourage greater communication between patients and physicians about cancer drugs approved on preliminary surrogate measures,” Liu said. “Although we are sure that physicians aspire to do this, it is important to keep in mind that once a drug is ‘approved’ by the FDA—especially under the accelerated approval pathway—this does not mean that a drug works well or better than other therapeutic alternatives. As always, a nuanced view of available evidence is critical to best serve patients.”
For example, Cliff added, “Some of these therapies have not been shown to extend life and I think patients should know that when they elect to pursue these therapies whether that is in a clinical trial or care setting.”
In 2022, the FDA’s Oncology Center of Excellence (OCE) launched Project Endpoint to evaluate the use of early, novel endpoints in oncology drug development and their relation to later endpoints such as overall survival. This included the “FDA-AACR-ASA Workshop: Overall Survival in Oncology Clinical Trials” in July 2023, in which the FDA, AACR, and the American Statistical Association (ASA) brought together stakeholders from across the drug development continuum to discuss the value of using overall survival as an endpoint as well as the difficulties involved in monitoring for it over the long term. On Saturday, during the AACR Annual Meeting 2024, the session “FDA’s Project Endpoint and Overall Survival in Oncology Clinical Trials” shared insights from that workshop. Anyone with and Educational Program Pass can watch the full session on the Virtual Meeting Platform.
Both the poster and the session were part of the Regulatory Science and Policy track at the AACR Annual Meeting 2024. Additional sessions on this track include plans from the FDA Center for Tobacco Products to prioritize the reduction of tobacco use, the efforts being made to increase diversity in oncology clinical trials, and an examination of how FDA regulators’ own experience as patients or caregivers has impacted their review process.

